
Device Labeling: Best Practices

Steps to ensure your medical device labels are in compliance with global regulations and have the longevity to withstand your device’s intended use.
Steps to ensure your medical device labels are in compliance with global regulations and have the longevity to withstand your device’s intended use.


Gain insight into the notified body approach to assessing biological equivalence and how manufacturers can make sure their equivalence justifications pass notified body review.


Per EU MDR regulations, any medical device on the market must be considered “state-of-the-art.” However, the term is not explicitly defined. Exploring MDR verbiage around standards harmonization, risk management and clinical data may allow a clearer understanding of regulatory expectations to emerge.


The specifications set uniform and rigorous benchmarks for tests across the EU, with the goal of clarifying the requirements for market actors and protecting EU patients.


The U.K. Medicines and Healthcare products Regulatory Agency (MHRA) has published its response to concerns raised by medical device trade associations regarding future regulation of medical devices in the United Kingdom.
The partnership brings together Evidence Partners’ literature review platform, DistillerSR, and Akra Team’s strategic regulatory services.
The partnership brings together Evidence Partners’ literature review platform, DistillerSR, and Akra Team’s strategic regulatory services.
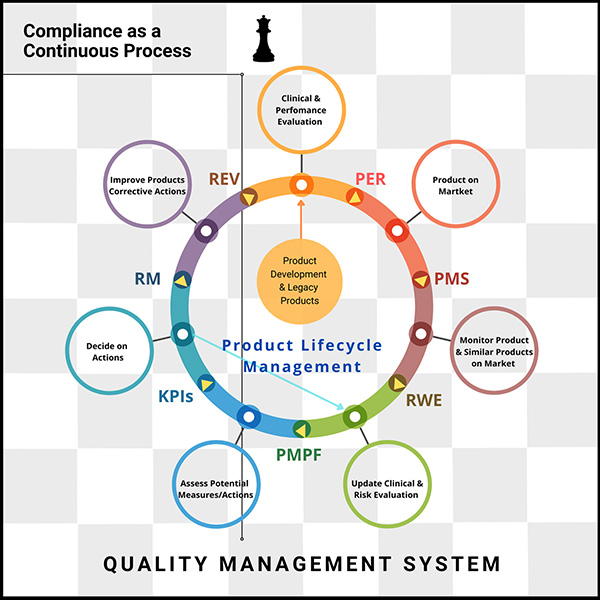
The only constant today is change itself.
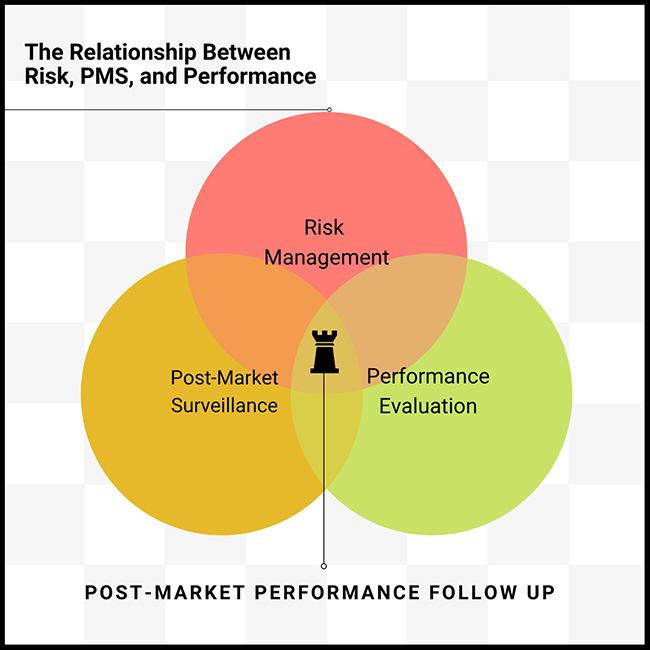
Understanding EU MDR and IVDR: The goal of EU MDR and IVDR is to ensure safety by asking manufacturers to provide evidence that their products are safe (disclosing any risks), effective (performing as expected), and state of the art (compared to industry benchmarks).


Embracing compliance is a continuous process, and investing in agile technologies that streamline workflows—especially in meeting EU MDR and IVDR requirements—is essential, says Lana Feng, Ph.D., CEO-founder of Huma.AI, a pioneer in a human-centered AI.


EU regulations require manufacturers of medical devices and systems prove the single-fault safety of their products. However, it is not clearly defined in detail how to comply with these requirements. This article explains which technical and legal requirements apply and which aspects should be considered during development.