The goal of EU MDR and IVDR is to ensure safety by asking manufacturers to provide evidence that their products are safe (disclosing any risks), effective (performing as expected), and state of the art (compared to industry benchmarks).
Furthermore, according to Martin King, a QA/RA medical device consultant who specializes in IVDR and EU MDR compliance, the regulations are designed to not only ensure that manufacturers comply with specific minimum standards but also to develop a fully transparent regulatory framework that encourages innovation–pushing companies to produce the highest quality, safest and most State of the Art products.
In other words, said King, “EU MDR and IVDR should help manufacturers to create ‘flawless’ devices.”
To that end, King said, “the regulations spell out exactly what manufacturers need to do in order to achieve perfect compliance, and indeed build flawless devices.” The main challenge is “not losing sight of the forest for the trees,” said King. EU MDR and IVDR are asking manufacturers not to leave any boxes unticked and, more importantly, make all their submissions entirely consistent,” said King.
When making submissions, said King, the question to consider is “What is it all about?” King said it’s about “the story, making the life of the reviewer as easy as possible, and being consistent in your storytelling all the way through all your documentation.”
Therefore, according to King, reviewers scrutinize submissions looking for inconsistencies, including technical documentation inconsistencies and the logical relationship or evidence presented to support the submission.
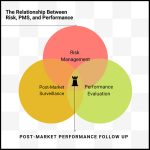
A Story of Risk, PMS, and Performance Evaluation
The logical connection between Risk, PMS, and Performance Evaluation
According to an IVDR eBook from MedTech Europe, Post-Market Performance Follow-Up (PMPF) is a continuous process that updates the performance evaluation. It shall be explicitly addressed in the manufacturer’s post-market surveillance plan.
However, according to Martin King, to truly comprehend the pivotal role of the PMPF, it’s essential to understand the logical relationship between Risk, PMS, and Performance Evaluation. As indicated in the diagram below, PMFP ties these three disciplines together:
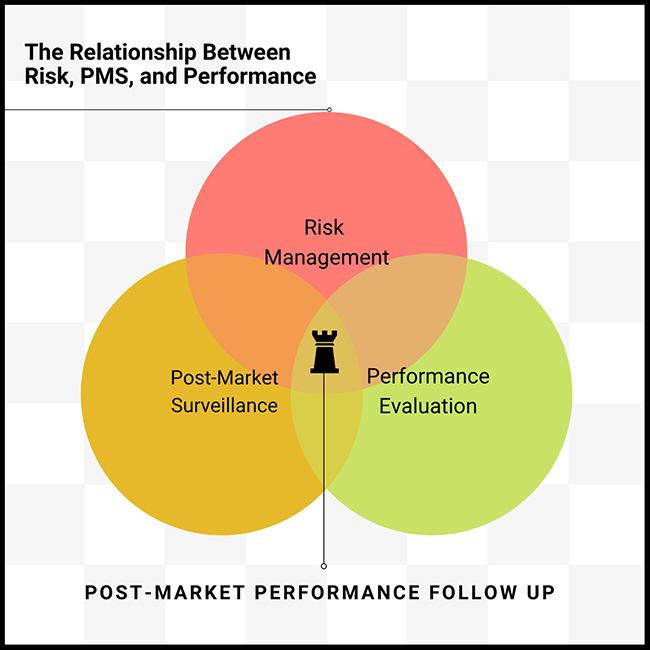
Risk Management: Is the device safe?
IVDR Section 3, Annex I – Requires manufacturers to establish, implement, document, and maintain a risk management system to identify and understand risks throughout the product life cycle.
Performance Evaluation: Can you prove it works?
IVDR Chapter VI – Requires manufacturers to establish a continuous performance evaluation process to ensure conformity with relevant general safety and performance requirements under the ordinary conditions of the intended use of the device, based on “sufficient data” establishing scientific validity, as well as analytical and clinical performance data.
Post-Market Surveillance: Does the device perform as expected?
IVDR Annex III – Requires the technical documentation on post-market surveillance to be drawn up and presented in a clear, organized, readily searchable, and unambiguous manner.
Additionally, IVDR Chapter VII requires manufacturers to plan, establish, document, implement, maintain and update a post-market surveillance system to actively and systematically gather, record, and analyze relevant data on the quality, performance, and safety of a device throughout its entire lifetime.
Post-Market Performance Follow-Up: How will you continue to monitor the device’s safety, performance, and scientific validity?
IVDR Annex XIII – Requires manufacturers to verify the device’s safety, performance, and scientific validity throughout its expected lifetime, ensuring the ongoing acceptability of the benefit-risk ratio and thereby detecting risks based on new evidence. 8
Why Are Post-Market Surveillance and Vigilance Necessary?
Understanding the difference between PMS and vigilance
According to Thema, Post-Market Surveillance (performed by the Manufacturer) is a proactive and systematic process designed to monitor the performance of a medical device by collecting and analyzing information relating to its use in the field. PMS is different from Vigilance (performed by the Manufacturer, the Authorised Representative EU REP, and sent to the competent Authority), which is a reactive process and consists in reporting serious incidents and Field Safety Corrective Actions (FSCA) to the Competent Authorities involved.
Both Post-Market Surveillance and Vigilance are necessary to identify gaps sooner, apply real-world data to reduce risk, and ideally eliminate flaws before they become problems.
Moreover, in a recent interview, Robert Maczejka, VP, Quality Management & Regulatory Affairs at Bloom Diagnostics, explained how Post-Market Surveillance is also a significant opportunity to reduce costs by monitoring standard KPIs such as complaints, nonconformities, and failure rates that systematically link to internal manufacturing KPIs.
“What PMS is telling us,” Maczejka said, “is there could be a problem.”
Post-Market Surveillance, if done thoroughly, can be an indicator of where to optimize products and prevent potential failures in the field, explained Maczejka. He noted that failures in production can often be contained with some minor adjustments, but when failures happen in the field, they are more costly to correct.
As Maczejka said, “The new cost chain is every dollar in R&D spending is worth $100 spent in production and is equal to $1,000 in customer success spending.” Therefore, he said, “PMS enables you to pull these economic levers when it’s not as costly.”
Furthermore, Maczejka stated, “The goal of all the regulations is, of course, safety. It’s not only safety but also the safety and the effectiveness for the patient. So, the better your job before releasing the product initially to market, the better the outcomes. Whereas, he said, “PMS is the key leverage point to optimize products that have already been brought onto the market.”
Another benefit of PMS, said Maczejka, is it can tell us if a product is useful to the users or not. Many products on the market are overloaded with more features than what users need. PMS provides an accelerated understanding of what customers are doing with products, identifies the essence of the products, and identifies where products may be over-engineered.
Therefore, “PMS is not only about gathering product feedback,” said Maczejka, “but it’s also about making perfect products, in multiple senses.”
* * *
This article is reprinted from “The Post-Market Gambit: After EU MDR and IVDR—A Systematic Compliance Framework for Driving MedTech Innovation,” Edited by Bassil Akra, Ph.D. Download the full 60-page eBook here for free or purchase it on Amazon.com.
Related Articles
-

The European Commission developed the new proposal following a December 9, 2022, meeting of the…
-
The partnership brings together Evidence Partners' literature review platform, DistillerSR, and Akra Team's strategic regulatory…
-
The only constant today is change itself.
-
On Monday, March 20, the EU MDR extension approved on March 7 came into effect…