
European Commission Proposes Extension for EU IVDR Compliance

If adopted, companies would have additional time—from 2027 to 2029, depending on the type of device—to gain approval under EU IVDR.
If adopted, companies would have additional time—from 2027 to 2029, depending on the type of device—to gain approval under EU IVDR.


IVDR has significant implications for the manufacturing of IVD devices, as it requires manufacturers to comply with new and more rigorous regulatory requirements. Wiktoria Banczyk, Product Manager Lab Filtration Medical Devices at Sartorius Lab Instruments, discusses the challenges posed by the implementation of EU IVDR 2017/746 and key considerations for manufacturers as they navigate today’s regulatory landscape.
The only constant today is change itself.
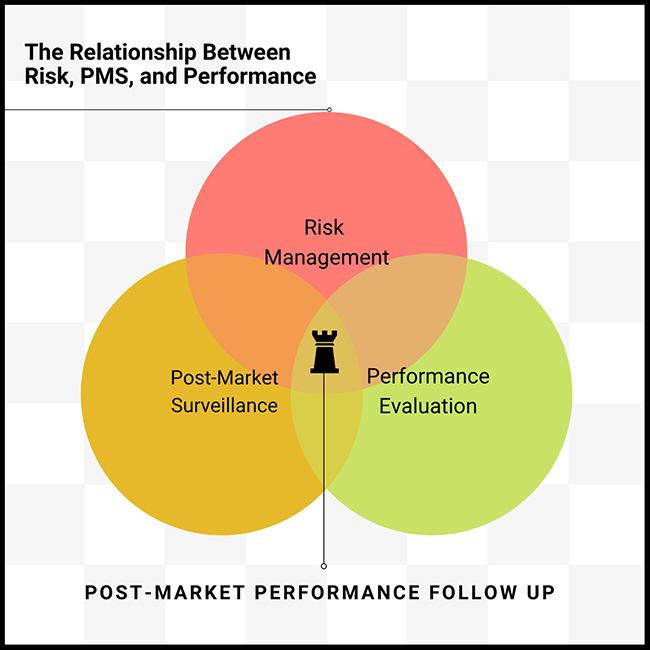
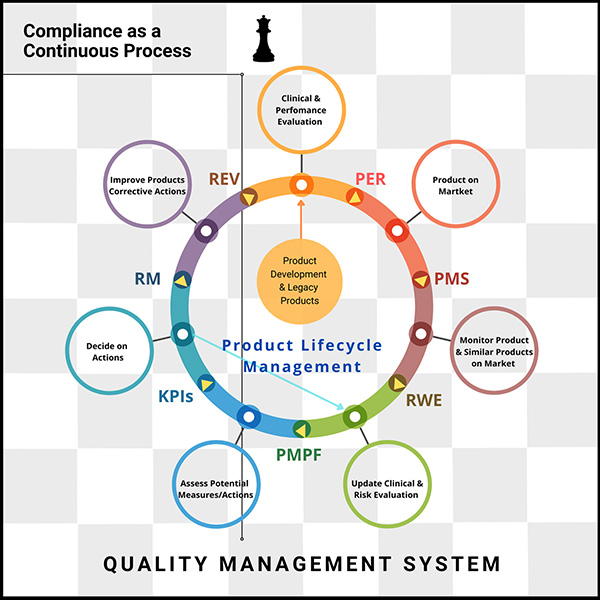
Understanding EU MDR and IVDR: The goal of EU MDR and IVDR is to ensure safety by asking manufacturers to provide evidence that their products are safe (disclosing any risks), effective (performing as expected), and state of the art (compared to industry benchmarks).


This article highlights what manufacturers should prioritize and areas in which new information may still emerge, along with providing a starting point for businesses that need to familiarize themselves with the UK Conformity Assessed marking requirements.


Medical devices and medical software are becoming increasingly connected to hospital networks, other medical devices or the Internet. As a result, manufacturers and developers are required to consider cybersecurity from the very early stages of development. This in turn necessitates comprehensive risk management along the entire lifecycle of a device.


The medtech regulatory environment in Europe has entered a new era. A Europe-wide medical device regulation has come into effect, presenting both challenges and opportunities for stakeholders in the sector.


A review of common risks and pitfalls of incorporating artificial intelligence in medical devices and an overview of the regulatory framework.


Beyond simply allowing for broader yet more efficient searches, leveraging software to automate literature reviews can organize references, assign screeners, and review screening decisions. This saves time, reduces bottlenecks, and, most importantly, leads to a highly transparent, standardized, and repeatable process that supports continuous CER and PER submissions across a product portfolio and for the life of a device.
Anyone can read the regulation. The challenge is in how to apply it to your company’s structure and product line.