In another first for warning letters, complaint handling supplanted CAPA as the violation found most in letters.1 Medical device manufacturers often struggle to keep up with complaint investigations and MDRs, but that’s not even their biggest challenge. The biggest challenge is establishing and maintaining a feedback loop from post-market surveillance to risk management, so that decisions about risk controls and future designs can be informed by actual market performance.
This article reviews the most important aspects of integrating the risk management and post-market surveillance processes, and review options in resolving some of the most common concerns. Part two of the article will discuss these aspects from clinical and engineering perspectives.
One of this article’s authors, Roberta Goode, will be speaking at MedTech Intelligence’s Integrated Complaints Management Conference, September 17-18, 2015. Register hereComplaint handling is a means of monitoring product performance in the field, post-commercialization or limited product release. It’s a critical step in monitoring, analyzing and evaluating risk in the clinical setting. In the development of a device, risks of similar and predicate products are documented as the basis for the estimation of risks associated with the new device, along with the use of clinical literature and expertise. In many cases, and especially for breakthrough or novel products, many risks are not known or are only vaguely known. In fact, all risk management is actually risk estimation. Actual severity and occurrences are unknown and can only be estimated by a cross-functional team of engineering and clinical experts. It is not until both a device and a procedure have been well-understood in clinical use that risk estimations can be considered reliable.
It is therefore imperative that medical device manufacturers have robust processes for monitoring complaints in the field and reviewing their risk estimations consistently, in order to:
- Identify new or previously unforeseeable hazards, hazardous situations and harms
- Determine whether risks are being maintained within acceptable levels
- Monitor the state-of-the-art to determine if risk has been reduced as low as possible
To do this in a compliant way, let’s start with the regulations that require risk management and complaint handling activities.
Complaint Handling Regulations
The requirements for complaint handling are fairly straightforward. In the United States, FDA’s 21 CFR 820 Subpart M, Records, addresses complaint files. 21 CFR 803 covers Medical Device Reporting. Outside of the United States, the European Commission’s MEDDEV 2.12-1 Guidelines on a Medical Devices Vigilance System, as well as ISO 13485 Section 8.1.2, provide comparable requirements.
Risk Management Regulations
In contrast, the requirements for risk management are difficult to find, and when located, they are challenging to interpret and implement.
In the United States, FDA’s 21 CFR 820, The Quality System Regulation (QSR), makes mention of risk analysis in Subpart C, Design Controls. This is currently the extent to which the FDA attempts to enforce risk management officially. However, we have witnessed several 483 observations for lack of adequate risk management in which FDA inspectors cited this element of the QSR as support.
More recent efforts by FDA to provide regulations for risk management include the guidance on human factors and quality by design.2,3 However, FDA lacks a complete guidance on the use of specific risk management tools for device manufacturers.
Outside the United States, ISO 14971:2007 is the international risk management standard.4,5 In order to meet the applicable Medical Device Directive, medical device manufacturers must conform to both ISO 14971:2007 and EN ISO 14971:2012.6 The release of EN ISO 14971:2012 to include Annexes ZA, ZB and ZC for clarification of the inherent alignment between risk management and the Medical Device Directive is critical to the proper application of risk management and complaint handling in medical devices, since it mandates the reduction of all risks as far as possible, considering state of the art and known stakeholder concerns. To conform to EN ISO 14971:2012, updates to existing product documentation may be necessary based on new guidance provided to industry through a Consensus Paper for Interpretation and Application of Annexes Z in EN ISO 14971:2012 version 1.1.7
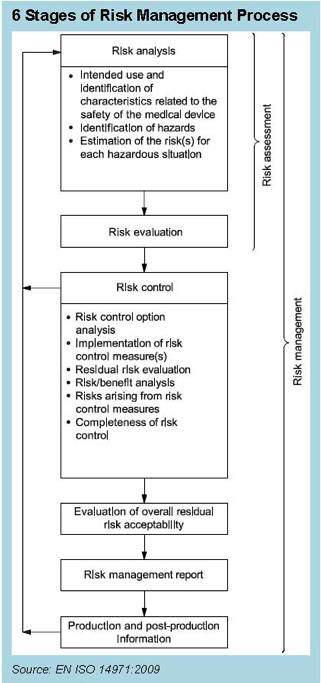
The Content Deviations contained in EN ISO 14971:2012 upend many long-standing practices in our industry. For example, no longer can device manufacturers count on instructions for use to reduce risk. That labeling must still exist, but it can no longer be used to mitigate risk in the FMEA. Furthermore, the concept of reducing risk as low as reasonably practicable (ALARP), which includes economic considerations, was replaced with a requirement to reduce risk as far as possible (AFAP), and the complexity of risk assessment and reduction becomes more complicated than ever. Beyond the technical challenges involved with risk assessment, there are the potential disparities between the benchtop product performance and clinical performance. It’s one thing to assesses expected risks in the product design phase, but reality in a clinical setting can be quite another. Therefore, it makes sense that post-market information must be fed back into the design of products. For that reason, the ISO 14971 standards reference the essential feedback loop from post-market information into risk management, as shown in Figure 1. The purpose of this feedback loop is to take the information from the complaint handling process and use it to review and revise the risk analysis in order to determine whether the benefit of the device continues to outweigh the risk, which requires addressing the three challenges previously mentioned:
- Identify new or previously unforeseeable hazards, hazardous situations and harms
- Determine whether risks are being maintained within acceptable levels
- Monitor the ever-changing state-of-the-art to determine if risk has been reduced as low as possible
Identify New or Previously Unforeseeable Hazards, Hazardous Situations and Harms
The core challenge in the identification of new hazards, hazardous situations and harms is garnering the right information and enough information from the end user in their complaint submission. The receipt of accurate information is critical in determining the true root cause(s) of the failure and making accurate risk-based decisions. Complete and accurate information can be difficult to obtain, for several reasons:
- Clinicians must report their perception of the apparent failure mode, which may or may not always align with engineering theory or terminology
- Complex reporting requirements can lead to the deliberate avoidance of complaint submission or a reduction in the provision of useful information
- Insufficient detail as it pertains to the hazardous situation that occurred is often provided, due to it being unknown or potentially legally implicating
- Insufficient detail as to the specific harms (if any) incurred by the patient is often provided, due to the terminology used having been too general
There are many options for minimizing lost information or missing information from your complaints reporting, which include:
- Simplifying your complaint forms as much as possible, and requiring only the mandatory fields, so as to reduce the burden to provide unnecessary information
- Asking clear and relevant questions in your complaints forms so as to target the right information and to improve the probability of receiving accurate information
- Collaborating with your sales team to follow-up on missing complaint data, which is a form of good customer service and provides the opportunity for in-servicing
- Providing options for clinician training, in order to consistently improve their level of knowledge of the device and any terminology that may be helpful in properly assessing the complaint data
Determine Whether Risks Are Being Maintained Within Acceptable Levels
In order to carefully assess device performance, manufacturers need to be able to perform several tasks well during complaint analysis, including:
- Achieving specificity about the patient population, intended use, and the clinical procedure being performed,
- Understanding details about the use scenario that occurred, including the user, the environment, and any ancillary devices or product interactions, to determine whether use-related failure modes may have contributed to the failure or whether there are usability issues with the device
- Ensuring consistent severity ratings for similar outcomes are available within the organization in the form of a severities listing
- Capturing the hazards and hazardous situations consistently, allowing for differentiation between “as reported” and “as analyzed” hazards
Once the organization has obtained these four critical pieces of information (patient, use scenario, severity, and hazard/hazardous situation), then the engineering team has the ability to produce educated feedback about the device’s performance and its associated risk, based on the information provided, as well as the investigation of the returned goods.
The first step in determining whether risk levels have increased or exceeded acceptable limits is to understand how the hazard and/or device failure mode occurred by using the hazardous situation and use scenario as context for the evaluation. Some questions to consider:
- Was the device used inside or outside of its intended use?
- Were the instructions for use properly followed in whole or in part?
- Was the hazard new or previously unforeseeable?
- Was the hazardous situation unique or commonplace?
- Was the use scenario unique or commonplace?
- Were all fluids, energies, devices, and tools that interacted with the device accounted for in the risk analysis?
These questions are used to determine the bounds of the investigation. Analysis of the product can then provide clues as to whether the failure was design-related, use-related or material-related.
In order to determine whether the severity should be increased: Based on the investigation, the hazard, its specific hazardous situation, and subsequent harm should be compared against the risk analysis to determine whether:
- The harm that occurred was previously identified
- The harm was more severe than previously anticipated (e.g., localized bleeding vs. severe bleeding)
In order to determine whether the occurrence should be increased: Based on the investigation, the manufacturer often makes a quantitative assessment based on the number of times a situation occurred relative to the overall opportunities for occurrence. However, it has been our experience that FDA is less interested in a quantitative evaluation of frequency of occurrence than in general, systemic trends towards increasing or decreasing frequency of occurrence. Qualitative assessments of risk may be more appropriate than quantitative assessments, as quantitative assessments are subject to minimization error due to multiplying serial fractions.
Monitoring State-of-the-Art to Determine if Risk Has Been Reduced as Far as Possible
Complaint Handling is a great way of monitoring. State of the art, which is defined in ISO 14971 to mean what is currently and generally accepted as good practice. It is somewhat subjective and determined by the combination of several different factors:
- The intended use and the indications for use for a specific procedure
- The specific technology being used
- Medical practice at the time of design (use and reasonably foreseeable misuse)
- Known stakeholder concerns, as seen through complaints
Since EN ISO 14971:2012 now requires the reduction of risk as far as possible, considering state of the art, it is important to be monitoring complaints for a change in state of the art. For example, during the review of complaint data, manufacturers may see an increase in complaints of a certain nature, not because the device has failed to function as intended, but because of clinicians’ changing:
- Perception of risk
- Tolerance of risk
- Preference for an alternative device or therapy, which they feel is safer
This analysis is subjective. However, it is critical that manufacturers consider all these factors, since it is the responsibility of the manufacturer, per EN ISO 14971:2012 content deviations, to reduce the risk as far as possible, considering state-of-the-art and known stakeholder concerns.
If the manufacturer were to see an increasing trend of complaints due to their product becoming less favorable in the market place, due to increasing safety concerns, they would have the obligation to implement a market correction and/or design changes to further reduce the risk to as low as possible.
Part II of this article, “Considerations When Using Postmarket Data in Risk Management“, reviews how to incorporate complaint handling and risk management into the postmarket surveillance.
References
- Schmitt, S.M., (March 14, 2013). Record Number of Warning Letters Issued in 2012; Complaint Handling Troubles Significant. The Silver Sheet, Article #09130313001.
- Food and Drug Administration. (June 22, 2011). Draft Guidance for Industry and Food and Drug Administration Staff – Applying Human Factors and Usability Engineering to Optimize Medical Device Design.
- Nasr, M.M. (February 28, 2007). “Quality by Design (QbD) – A Modern System Approach to Pharmaceutical Development and Manufacturing – FDA Perspective”. FDA Quality Initiatives Workshop. North Bethesda, MD.
- International Organization of Standardization. (March 1, 2007). ISO 14971:2007 Medical devices – Application of risk management to medical devices.
- International Organization of Standardization. (July 31, 2012). EN ISO 14971:2012 Medical devices – Application of risk management to medical devices.
- European Commission. Council Directive 93/42/EEC of 14 June 1993 concerning medical devices.
- European Association for Medical devices of Notified Bodies. (October 13, 2014). Consensus Paper for the Interpretation and Application of Annexes Z in EN ISO 14971:2012. Version 1.1.
*Figure 1 credit: This excerpt is adapted from ISO 14971:2007, Figure 1 on page 6, with the permission of ANSI on behalf of ISO. (c) ISO 2015 – All rights reserved.
Related Articles
-

Mitigating risk of failure is one of the most important disciplines to perform for medical…
-

Bringing design and quality assurance processes together earlier in the device development process can reduce…
-

Both legacy players and startups have an important role to play when it comes to…
-

The acquisition is expected to strengthen PTC’s closed-loop product lifecycle management offerings by extending the…