

Gain insight into the notified body approach to assessing biological equivalence and how manufacturers can make sure their equivalence justifications pass notified body review.
Gain insight into the notified body approach to assessing biological equivalence and how manufacturers can make sure their equivalence justifications pass notified body review.

Before product developers can more confidently design RWE studies and venture away from the traditional path of lengthy prospective studies, industry must overcome several challenges. This article provides insight on these barriers and perspective on how industry might fully realize the promise of RWE in the near-term to advance health care.

“Approaches to Increasing Diversity in Clinical Research and Addressing Health Inequities” offers key recommendations for device manufacturers and developers on steps they can take to increase diversity in clinical research and address health equity.

Modern technology has given rise to new legal questions. How does FDA regulate machine-learning computers that are changing so rapidly – given that the approved product may be drastically different than the product that ends up on the market? These questions arise from a lack of understanding of the complex nature of AI/ML-based SaMD, the opaqueness of the regulatory framework, and a dearth of relevant case law.

The Medical Device Regulation (EU) 2017/745 became the applicable EU law on May 26, 2021. After a four-year transition period, is anyone ready? Well, let us have a look.

The agency action includes all non-NIOSH-approved disposable respirators, including imported KN95s.

FDA also supports the removal of the HeartWare HVAD System from the market.
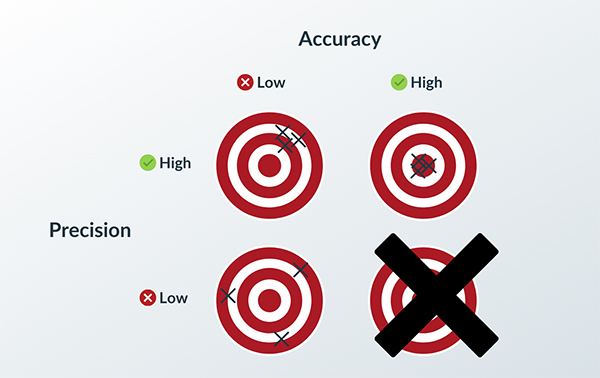
The key is to keep quality high and risk low.

It’s time for medtech design engineers to take a page from the enterprise security playbook.

The goal is to establish a baseline of cybersecurity hygiene and assurance for devices that are part of the national critical infrastructure software supply chain.





