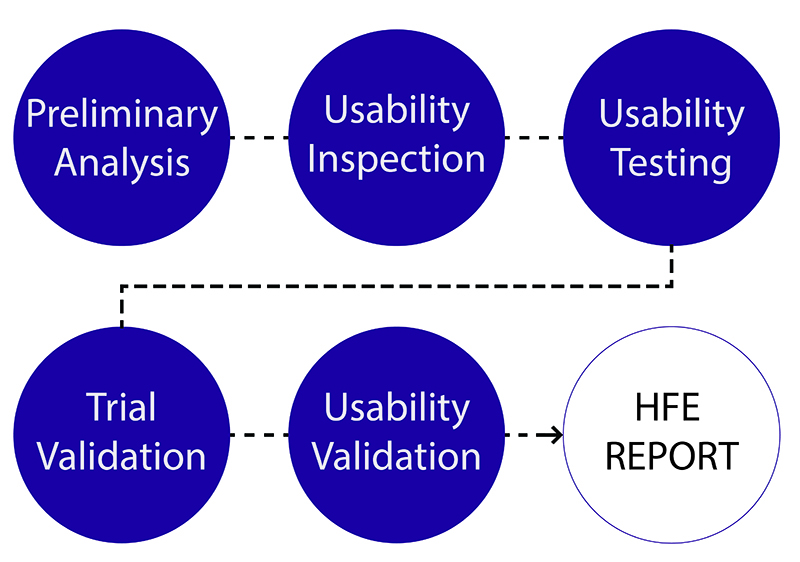
Last week FDA issued the long-awaited final guidance on human factors and usability, targeting the device user interface—from displays and controls to instructions for use, labeling and packaging. The guidance, Applying Human Factors and Usability Engineering to Medical Devices, will supersede the nearly 16-year-old guidance, Medical Device Use-Safety: Incorporating Human Factors Engineering into Risk Management. “CDRH believes that for those devices where an analysis of risk indicates that users performing tasks incorrectly or failing to perform tasks could result in serious harm, manufacturers should submit human factors data in premarket submissions (i.e., PMA, 510(k)),” the guidance states. As such, CDRH released a draft guidance, List of Highest Priority Devices for Human Factors Review, which details the following devices that should include human factors data in premarket submissions:
- Ablation generators
- Anesthesia machines
- Artificial pancreas systems
- Auto-injectors
- Automated external defibrillators
- Duodenoscopes with elevator channels
- Gastroenterology-urology endoscope ultrasound systems with elevator channels
- Hemodialysis and peritoneal dialysis systems
- Implanted infusion pumps and external infusion pumps
- Insulin delivery systems
- Negative pressure wound therapy (intended for home use)
- Robotic catheter manipulation systems
- Robotic surgery devices
- Ventilators
- Ventricular assist devices

“FDA believes these device types have clear potential for serious harm resulting from use error, and that review of human factors data in premarket submissions will help FDA evaluate the safety and effectiveness and substantial equivalence of these devices,” the document states. The draft guidance is open for comments until May 3, 2016.
The agency addressed human factors in combination products in a draft guidance released last week. Human Factors Studies and Related Clinical Study Considerations in Combination Product Design and Development defines human factors concepts and studies, and outlines the process considerations for submitting human factors study data for combination products. It recommends data should be submitted for products intended for use outside the healthcare environment, including products that require self-administration by patients, and combination products that have a device constitute part for which human factors data should be submitted. If the device does not fall into either of these segments, the agency recommends performing risk analysis to assess the need for human factors studies. The draft guidance, which is open for comments until May 3, 2016, reviews considerations for design changes after human factors validation as well as investigational applications, and the relationship between human factors and major clinical studies.
CDRH also released a draft guidance on NHRIC or NDC numbers on device labels and packages, Enforcement Policy on National Health Related Item Code and National Drug Code Numbers Assigned to Devices. The agency is accepting comments for the next 60 days.
Related Articles
-

“Content of Human Factors Information in Device Marketing Submissions” and “Voluntary Malfunction Summary Reporting (VMSR) Program for Manufacturers” are now open for comment.
-

By applying HFE principles early on, packaging design can evolve beyond usability evaluations in product development cycles to ultimately ensure a better user experience and safe execution.
-
Hägen, who was a contributor to Medtech Intelligence, spent 28 years building BlackHägen Design to support clients in the development of “mission-critical” products. He was pivotal in the advocacy of human factors and good design practices for medical devices and…
-

The MDDT program was developed in collaboration with the National Institutes of Health’s (NIH’s) National Institute of Drug Abuse (NIDA), National Institute of Dental Craniofacial Research (NIDCR) and National Cancer Institute (NCI) as a way for the FDA to qualify…