
Medical devices come in all shapes and sizes, ranging from wheelchairs to cardiac pacemakers and everything in between. All medical devices must meet the requirements of the EU Medical Device Directive 93/42/EEC (MDD) in order to be CE marked and placed on the European market. However, when a medical device incorporates a drug, some additional specific requirements must be met.
Achieving certification can be a complex, time-consuming and costly process. These frustrations and delays can be mitigated by enlisting the right resources and product development team with expertise in creating a realistic time-to-market strategy based on knowledge of each stage of the drug–device regulatory process.
Is the Product Regulated as a Medical Device or a Drug?
Determining the correct regulatory strategy for a drug-device combination product can be a complex task. As a first step, consideration must be given to the European regulations’ definitions of both a medical device and a drug.
According to Article 1 of the EU Medical Device Directive 93/42/EC definition: “‘medical device’ means any instrument, apparatus, appliance, software, material or other article, whether used alone or in combination, including the software intended by its manufacturer to be used specifically for diagnostic and/or therapeutic purposes and necessary for its proper application, intended by the manufacturer to be used for human beings for the purpose of:
— diagnosis, prevention, monitoring, treatment or alleviation of disease,
— diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap,
— investigation, replacement or modification of the anatomy or of a physiological process,
— control of conception,
and which does not achieve its principal intended action in or on the human body by pharmacological, immunological or metabolic means, but which may be assisted in its function by such means.”
Article 1 of the EU Medicinal Products Directive 2001/83/EC defines a medicinal product as: “(a) Any substance or combination of substances presented as having properties for treating or preventing disease in human beings; or (b) Any substance or combination of substances which may be used in or administered to human beings either with a view to restoring, correcting or modifying physiological functions by exerting a pharmacological, immunological or metabolic action, or to making a medical diagnosis”.
The principal mode of action of the drug-device product is paramount in determining whether the product is regulated as a drug or as a medical device. Medical devices achieve their principal intended action through physical or mechanical means; however, they may be assisted by pharmacological, immunological or metabolic means. Drugs achieve their intended action through the pharmacological, immunological or metabolic action of a molecule.
There are two types of medical devices that incorporate a drug. The first type are products that are designed to deliver a drug, are supplied with the drug, and are not reusable (e.g. pre-filled insulin syringes). Under the EU Medicinal Products directive, these types of products are regulated as drugs. A medical device that incorporates a substance, which if used separately, is classified as a drug and is the second type. If the action of the drug is ancillary to that of the device (e.g. drug-eluting stent, bone cement containing an antibiotic), it is regulated as a medical device.
Key factors to consider when determining if the action of the drug is “ancillary” include:
- How does the product achieve its intended purpose?
- What is the primary mode of action?
- Liability of the drug to act on the human body
Examples of products that are regulated as medical devices and contain an ancillary drug include:
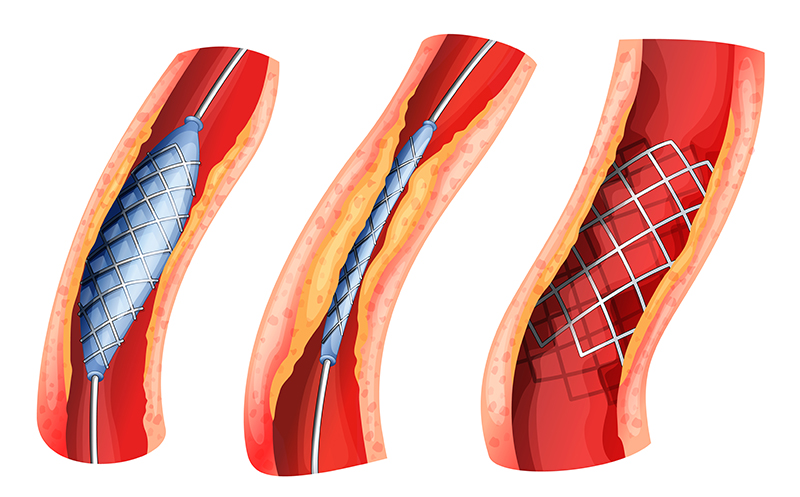
Drug-eluting Stents. Drug-eluting stents are placed in diseased peripheral or coronary arteries. A stent is typically composed of a metal or polymer mesh that is inserted by percutaneous coronary intervention, a procedure that widens the blocked artery by temporarily inserting and inflating a tiny balloon. The stent achieves its intended medical purpose through physical means: The scaffold keeps the artery open, while a drug coating prevents cell proliferation (i.e., growth of scar tissue inside the lining of the artery). As the primary mode of action is through physical means rather than through the action of the drug, the product is regulated as a medical device.
Wound Care Dressings. Another example is wound care dressings that incorporate silver. Silver sulfadiazine is classified as a drug and may be incorporated into wound care dressings for its antibacterial properties. However, the primary mode of action of a wound care dressing is physical (i.e., the absorption of exudate from the wound). Protection of the wound is achieved by physical means through use of the dressing. The antibacterial action of silver is considered to be ancillary to that of the wound dressing.
Classification
A medical device that incorporates an ancillary drug substance is classified as Class III, in accordance with Annex IX, Rule 13 of the MDD. This is the highest medical device risk classification, with the presence of the drug component adding a greater amount of complexity to the CE marking approval process.
It is critical to establish the correct classification of a medical device that incorporates as drug at the earliest possible stage. There are useful guidance documents freely available from the European Commission website and regulatory agencies such as the Medicines and Healthcare Products Regulatory Authority (MHRA) in the UK and the Health Products Regulatory Authority (HPRA) in Ireland.1-6 In addition, the MHRA and HPRA have processes whereby scientific advice can be sought to make a determination on the product classification.
CE Certification Process
Class III medical device manufacturers typically use the “Full Quality Assurance” procedure set out in Annex II of the MDD to obtain CE certification. Under this procedure, a design dossier must be compiled to show evidence of conformity with the Essential Requirements described in Annex I of the MDD. The design dossier must be reviewed by a Notified Body and a quality audit of the medical device manufacturer must be conducted by the Notified Body.

Where a medical device incorporates an ancillary drug substance, the regulatory process is somewhat more complex. The Notified Body must consult a Competent Authority before taking a decision on providing CE Mark certification. The manufacturer must compile a comprehensive dossier clearly describing the quality, safety and usefulness of the drug substance itself, and also on the drug as incorporated into the finished device. The dossier for the drug product is created in accordance with the common technical dossier (CTD) format. This CTD format is a mandatory format for new drug applications in the EU. This provides a regulatory challenge to medical device manufacturers, as specific expertise in drug dossier submission requirements is needed. The drug dossier is categorized into the following sections in Figure 1.
Table I includes the main dossier elements needed for this type of submission.
CTD Module |
Description |
| CTD Module 3 – Quality | Drug Substance section: Describes the characteristics of the drug substance, the manufacturing process and the controls applied to the drug substance.
Drug Product section: Describes the materials used, the method of manufacture and controls applied to the medical device incorporating the drug substance. The manufacturing process and analytical controls must be appropriately validated to international standards. |
| CTD Module 4 – Pre-Clinical | Toxicity, pharmacokinetics and local tolerance data to be provided. |
| CTD Module 5 – Clinical | Clinical data to be provided to address safety and usefulness of the medical device incorporating the drug. |
|
Table I |
|
Drug Consultation Process
Selection of a Competent Authority is a key step in the regulatory process. The manufacturer can choose the Competent Authority to perform the drug dossier review. Key deciding factors in deciding on a Competent Authority is experience in the review of similar drug molecules, and their willingness to be involved in this process. The Notified Body must consult the Competent Authority in writing, ideally six months before the intended submission to the Competent Authority. A pre-submission meeting between the manufacturer, Notified Body and Competent Authority is normally held approximately two months before the anticipated submission date. The Competent Authority will assess the dossier and advise of any reservations or outline the main areas of concern to the Notified Body. The Competent Authority issues their decision within six months. However, it should be noted that a “stop-clock” system is permitted whereby the Competent Authority will stop the clock for a given period once they have assessed and issued their list of deficiencies. Additional time should be allowed for addressing Competent Authority questions and deficiencies in the overall regulatory strategy of the product. The Notified Body will only issue the CE certificate when all elements are closed.
The additional drug consultation procedure significantly increases the time, cost and complexity involved in the product approval process of a medical device that incorporates an ancillary drug substance. It is advisable for medical device manufacturers to obtain support in defining the regulatory strategy for such devices at the earliest opportunity. Considerations such as correct device classification, selection of a certified drug supplier and site of manufacture, selection of an experienced Notified Body and Competent Authority, realistic timelines for drug dossier submission, and specific testing requirements for drug substances must be taken in to account.
There are inherent risks with the incorporation of a drug substance with a medical device, which clearly makes CE certification more complicated. The issue of patient safety is a critical consideration throughout this process. Each time a new medical device product is manufactured, there must be a high degree of confidence that the product can be used safely in a patient—with minimal or no risk. With the amount of effort that goes into a drug-device submission, it is incumbent on all the participants in the certification process to assure that medical devices that are used by patients and the general public are not only effective, but safe.
References
- European Commission. (June 2010). Medical Devices: Guidance Document, Classification of Medical Devices, MEDDEV 2.4/1 Rev 9. Retrieved from http://ec.europa.eu/growth/sectors/medical-devices_old/documents/guidelines/files/meddev/2_4_1_rev_9_classification_en.pdf
- European Commission. (March 2010). Borderline products, drug-delivery products and medical devices incorporating as integral part, an ancillary medicinal substance or an ancillary human blood derivative, MEDDEV 2.1/3. Retrieved from http://ec.europa.eu/growth/sectors/medical-devices_old/documents/guidelines/files/meddev/2_1_3_rev_3-12_2009_en.pdf
- European Commission. (July 2014) Manual on borderline and classification in the community. Regulatory framework for medical devices. Retrieved from http://ec.europa.eu/health/medical-devices/files/wg_minutes_member_lists/borderline_manual_ol_en.pdf
- MHRA. (June 2013). Borderlines between medical devices and medicinal products. Retrieved from https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/284493/Borderlines_between_medical_devices_and_medicinal_products.pdf
- MHRA. (November 2012). A guide to what is a medicinal product. MHRA Guidance No. 8. Retrieved from https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/398998/A_guide_to_what_is_a_medicinal_product.pdf
- HPRA. (April 27, 2009). Guide to classification of a medical device. HPRA Guidance ADV-G0004-2. Retrieved from http://www.hpra.ie/docs/default-source/publications-forms/guidance-documents/adv-g0004-guide-to-classification-of-a-medical-device-v2.pdf?sfvrsn=4
Related Articles
-

The UK MHRA has published “Software and Artificial Intelligence as a Medical Device.” The guidance…
-
“Addressing unmet needs across pediatric populations is critical to advancing children’s health, and we are…
-

The MDDT program was developed in collaboration with the National Institutes of Health’s (NIH’s) National…
-

More and more, the quality of real-world evidence (RWE)—gathered from remote patient monitoring data, product…