

Unique Device Identifiers (UDI) are unique numeric or alphanumeric codes that identify medical devices sold in the United States from manufacturing through distribution to patient use for the benefit of industry, FDA, consumers, healthcare providers and healthcare systems.
UDIs allow for more accurate reporting and analyzing of adverse events reports, reduce medical errors by enabling precise identification of a device, provide a standard documentation of device use in electronic health records, clinical information systems, claim data sources and registries, which will ultimately improve postmarket surveillance, and simplify medical device recalls. Once obtained, UDI information must be submitted to the Global Unique Device Identification Database (GUDID).
What is a UDI?
UDIs include the device identifier, which is fixed information including only the device labeler and the specific version or model of a device. UDIs may also contain production identifier information such as the lot or batch number within which a device was manufactured, its serial number, expiration date and the date the device was manufactured. Additionally, if the product is for human cell, tissue, or cellular and tissue-based product (HCT/P) regulated device, it must also have a distinct identification code as required by §1271.290(c).
Once fully implemented, FDA’s UDIs are expected to lead the development of a globally recognized medical device identification system.
By When Must I Comply?
FDA has been phasing in enforcement dates for UDIs with the next, September 24, 2020, as the date by which all “Class I devices, and devices that have not been classified into class I, class II, or class III that are required to be labeled with a UDI, must a bear UDI as a permanent marking on the device itself if the device is a device intended to be used more than once and intended to be reprocessed before each use. § 801.45.”1
Enforcement of UDI labeling (21 CFR 801.20 & 801.50), GUDID Data Submission (21 CFR 830.300), and Standard Date Format (21 CFR 801.18) requirements will begin September 24, 2021.
It is important to note that Class I or unclassified implantable, life-supporting or life-sustaining devices must already be in compliance with UDI requirements. Exemptions include Class I CGMP-exempt devices per 21 CFR 801.30(a)(2). Additionally, finished class I and unclassified devices manufactured and labeled before September 24, 2018, are exempted from the UDI labeling and GUDID submission requirements until September 24, 2021 per 21 CFR 801.30(a)(1).1
How Do I Get a UDI?
UDIs are issued by FDA-accredited issuing agencies which are permitted to assign UDIs according to the Unique Device Identification System final rule.
Once obtained, device labelers are required to submit basic device identifier elements for each device, (not production identifier) to serve as key device information in the database. In order to submit information to the GUDID, the device labeler must first request a GUDID Account.
UDIs must be applied on device labels, device packages, and in some instances, directly on the device. If a device is intended for more than one use and intended to be reprocessed before each use, the device labeler must also mark the UDI directly on the device.
UDIs must be in two forms on labels and packages, easily readable plain-text and machine-readable form that uses automatic identification and data capture (AIDC) technology. Figure 1 is an example from FDA’s website.
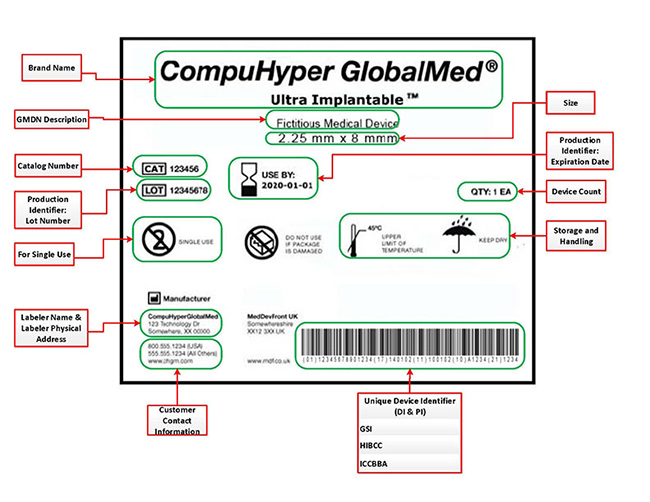
If you are a device labeler, or the company that applies the label to a device, or who causes the label of a device to be modified, with the intent that the device will be commercially distributed without any subsequent replacement or modification of the label, you must obtain a UDI and submit that information to GUDID no later than September 21, 2020 (with noted exceptions). In most instances, the labeler is the device manufacturer, but the labeler may be a specification developer, a single-use device reprocessor, a convenience kit assembler, a repackager, or a relabeler.
Now is the time to begin thinking about the process of obtaining UDI numbers for your devices and getting those numbers registered with GUDID. If you are unsure whether you are considered a labeler in the eyes of FDA, or need assistance with understanding your UDI requirements or obtaining a UDI number and submitting to GUDID, contact a reputable consulting firm for assistance. If you are a foreign firm intending to export devices to the U.S. market, notify FDA of your U.S. Agent.
Accurate and timely compliance with FDA’s law ensures your products will not considered misbranded, leading to FDA enforcement actions.
Reference
- FDA. “Compliance Dates for UDI Requirements”. Retrieved from https://www.fda.gov/medical-devices/unique-device-identification-system-udi-system/compliance-dates-udi-requirements.
Related Articles
-

The MDDT program was developed in collaboration with the National Institutes of Health’s (NIH’s) National Institute of Drug Abuse (NIDA), National Institute of Dental Craniofacial Research (NIDCR) and National Cancer Institute (NCI) as a way for the FDA to qualify…
-
The guidance is intended to clarify the FDA’s approach for referencing the terms “device” and “counterfeit device” in FDA documents, as well as how the agency intends to interpret existing references to section 201(h) of the FD&C Act in guidance,…
-

The Food and Drug Omnibus Reform Act (FDORA) authorized FDA to conduct remote device facility inspections. While it will take time for the FDA to take the necessary steps to start performing remote inspections, industry can look to remote regulatory…
-

The position paper from the European Commission’s Medical Device Coordination Group (MDCG) recommends an extended timeline to allow certain MDD or AIMDD certified legacy devices to come into compliance with MDR.