
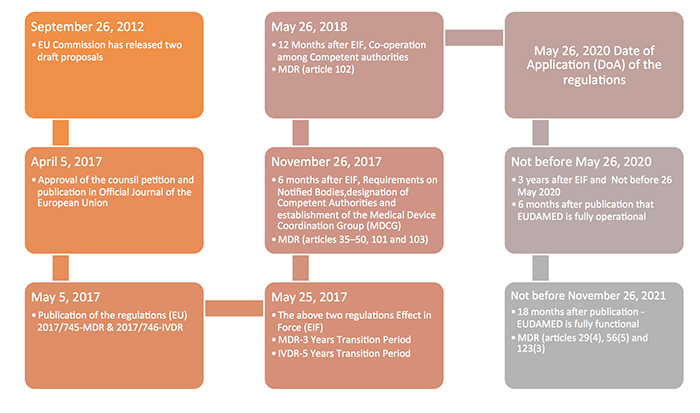
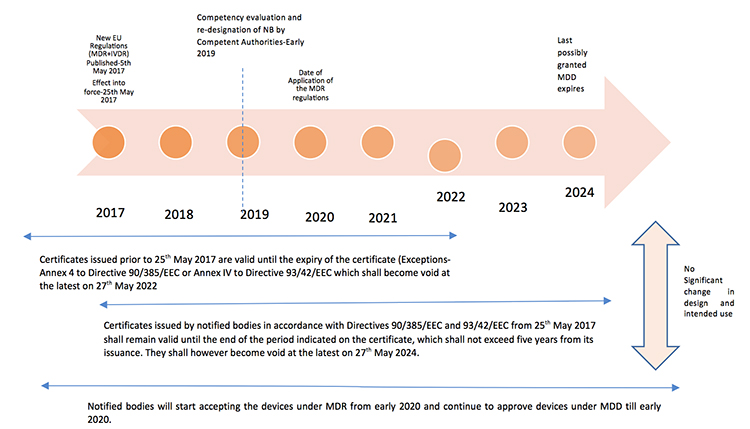
The new Europe (EU) Medical Device Regulations (MDR) published by the European Commission on May 5, 2017 revamped major portions of the EU Medical Device Directive (MDD), raising compliance bars for all device manufacturers, economic operators and notified bodies. The new regulations show a way forward towards the globalization of medical device regulations, which contribute to a high level of safety and facilitate easy trade across the borders by the introduction of unique device identification(UDI), general safety and performance requirements, technical documentation, classification rules, conformity assessment procedures and clinical investigations.
The term ‘medical device’ encompasses a variety of products, ranging from simple thermometers to artificial intelligence software programs for diagnosing diseases using patient test report data. Development of new products to meet user needs is progressing unimaginably with the advent of digitization and rapid evolution in medical and scientific innovation. Yet with the advent of serious cases involving devices such as the PIP breast implant scandal, there is a need for more stringent regulations.
Overview
“The current Regulatory approach which includes supervision of notified bodies, conformity assessment procedures, clinical investigations and clinical evaluation, vigilance and market surveillance should be reinforced to strengthen further. Also, more provisions should be introduced to ensure transparency and traceability regarding medical devices, to improve health and safety.” –Regulation 2017/745
| Medical Device Directive | Medical Device Regulations |
| 93/42/EEC (MDD)-43 pages 90/385/EEC (AIMD)-20 pages |
Regulation EU 2017/745-177 pages Repealing Council Directives 90/385/EEC and 93/42/EEC (MDD+AIMD) |
| 23 Articles, XVII Annexes | 123 Articles, XVII Annexes |
| Total number of rules for classification: 18 | Total number of rules for classification: 22 |
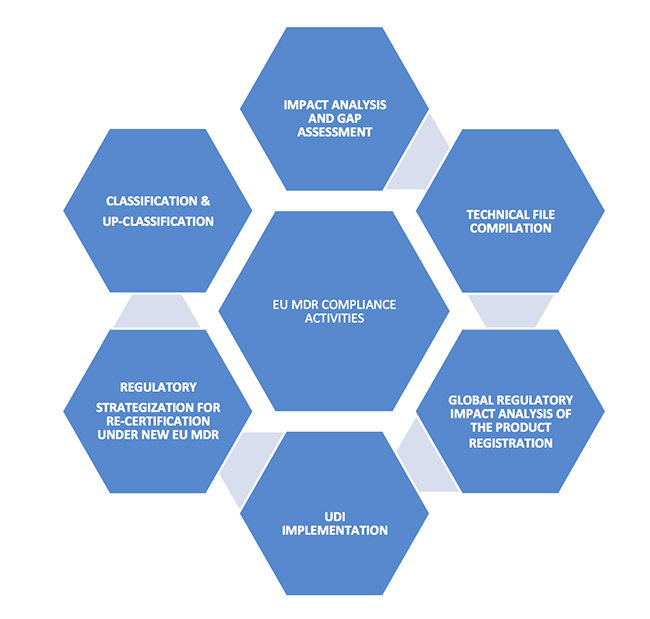
Understanding the MDR
1. Consider the classification of your device; finalization of your product portfolio
If your device is currently an accessory to a medical device, a product with aesthetic or another non-medical purpose, is a borderline product containing medicinal substances, or is a combination product, or software product, then the new medical device regulations should be evaluated for the classification and up-classification of the device.
The classification and up-classification of devices has a major impact on manufacturers, as EU MDR imposes tighter compliance, safety and efficacy requirements. This will play a key role in finalizing current and future product portfolios due to the significant cost of compliance. Any changes should start at the management level and include key decision makers who determine the product portfolio for the European market, assessing the impact of the global product approvals and market access based on CE certifications.
Non-compliance to the new requirements within the stipulated timelines will lead to loss of license to market your products in Europe. 2. Identification of the major compliance requirements
2. Identification of the major compliance requirements
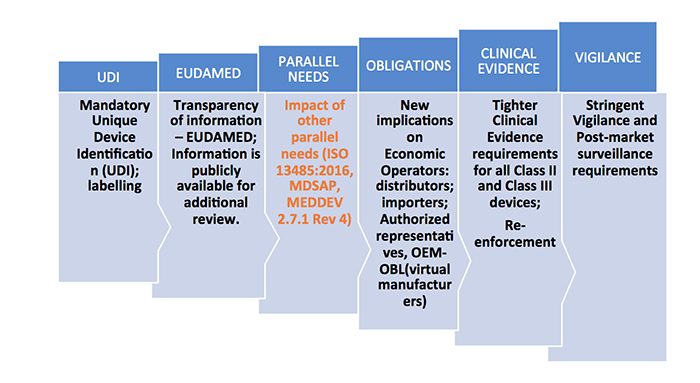 Familiarization of New MDR Terminology: Medical device co-ordination group (MDCG), General Safety and Performance Requirements, Common Specifications, Unique Device Identification, Virtual Manufacturer.
Familiarization of New MDR Terminology: Medical device co-ordination group (MDCG), General Safety and Performance Requirements, Common Specifications, Unique Device Identification, Virtual Manufacturer.
For the products that are newly classified as medical devices as per the new rules, review of the notified bodies will be heightened. New documentation should be made ready for CE certification as per the new regulations for both reclassified and existing products. Some of the key technical documents that should be in place include:
- Risk analysis documents
- Clinical data availability analysis and plan
- General safety and performance requirements
- Common specifications derivation
- All relevant QA SOPs
- Performance evaluation tests of the device as per the claims
- Labeling updates
- Alignment to the ISO 13485 requirements for manufacturing of the product and certification
- PMCF plans
Continue to page 2 below.
Related Articles
-

The position paper from the European Commission’s Medical Device Coordination Group (MDCG) recommends an extended…
-

The UK MHRA has published “Software and Artificial Intelligence as a Medical Device.” The guidance…
-
On Monday, March 20, the EU MDR extension approved on March 7 came into effect…
-
“Addressing unmet needs across pediatric populations is critical to advancing children’s health, and we are…