Your CAPA System: Operate Effectively and Maintain Profitability

The medical device industry faces the challenge of delivering safe and cost-effective products on time, every time. The pressure to introduce products into the marketplace is undoubtedly intensified by the strict guidelines enforced by the FDA and other global regulatory bodies. Medical device companies are obligated to meet market demands while fully complying with regulatory requirements.
Over the last several years, there has been a significant increase in audit findings, warning letters and consent decrees that have, in some cases, caused a supply chain delay or even plant closure. Every organization understands that an effective corrective and preventive action (CAPA) program is mandatory for doing business while still satisfying FDA and international regulatory guidelines. The bottom line is to find ways to operate effectively within a CAPA system while maintaining profitability.
Death by CAPA
While working with FDA district directors of both the Southern California and San Francisco districts several years ago and examining the device industry, one director stated that if we didn’t do something to move companies toward better risk management of the myriad inputs to the CAPA system they would suffer “Death by CAPA.” That simple comment went viral and is now commonly used throughout the industry.
In short order, we began to reexamine the CAPA “feeders” for the types of outcomes that were best suited to solve particular inputs. Better definitions were determined for corrections, corrective actions, and preventive actions. The front end of the CAPA process was replaced with a risk gateway that determined what to do based upon the severity of the situation and the probability of occurrence. At the same time, the work that we were doing with the Global Harmonization Task Force (GHTF) in an effort to harmonize a global standard reflected this needed change.
No longer is it appropriate to drive every quality event (complaint, deviation, product non-conformance, audit finding, etc.) into a one-to-one path toward a corrective or preventive action. Many quality events are handled through a correction or containment action and withdrawn from the more rigorous formal quality event management/CAPA process early.
Risk models can be developed to reflect the business of that device manufacturer that will eliminate many of the pains caused by Death by CAPA syndrome (See Figure 1).
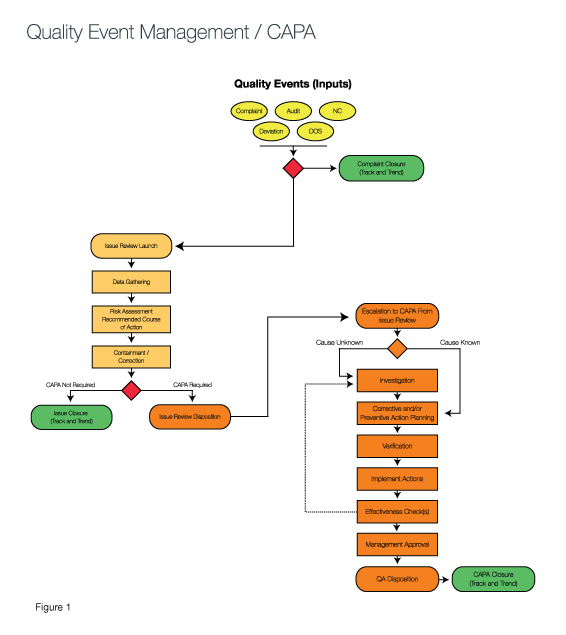
The green ovals in Figure 1 demonstrate how exit points in the process allow for quality events to be resolved at the simplest stage based upon the risk inherent to the event. Those that require a corrective action are investigated inside the CAPA process, while more simple events are resolved immediately with containment strategies or are monitored via tracking and trending until an action is needed. (Compare this to the GHTF/IMDRF Study Group Three Final Document, pages 5-6.)
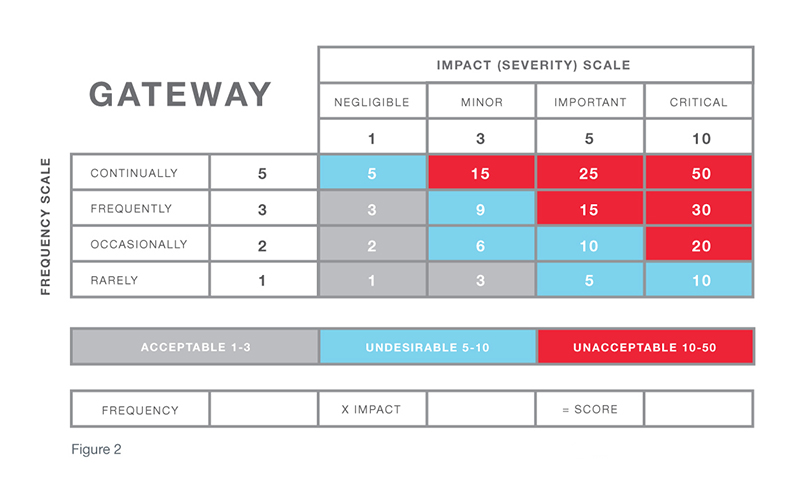
The risk assessment called out in the center section of the diagram is often referred to as the CAPA gateway. In a sense, by responding to the questions raised via the CAPA gateway and determining the associated risk, you understand the “finish line” required for each quality event. For example, if a quality event that has a risk factor in which the impact of the occurrence is serious and the frequency is high, an investigation is needed to determine root cause. Then, a corrective action must be taken to eliminate reoccurrence of the cause. At that point, verification is required to ensure the problem has been solved.
It is also wise to connect the corrective action to the change control process. Doing so makes it easier to determine if the change will result in potential problems either downstream in the process or as it may connect to the solution. These potential problems can be addressed with preventive actions that keep the potential problem from occurring.
Medical Device Case Study: One CAPA System, Two Quality Events
To better understand the benefits of risk gateways, consider the following example. A company that manufactured diabetes test strips had many quality events logged into the CAPA system. Two particular complaints from the field warrant deeper analysis:
- The ink color on the outside of the container was the wrong Pantone color of green (easily readable with no other errors)
- The test strip had caused a false positive reading and insulin was misjudged resulting in hospitalization (serious medical intervention)
The first example requires either no action or a correction. The second quality event requires a thorough investigation and corrective action. A risk gateway early in the CAPA process would have determined the appropriate finish line and required actions based upon risk assessment.
Unfortunately, the company had placed both events into the formal CAPA process and was then required to follow all the steps in the process—investigation, correction and validation—before the actions could be closed and the correct finish lines crossed. A CAPA process with exits similar to those shown in the process in Figure 1 would have allowed for an earlier exit with containment/correction rather than an exit later in the process when more rigor is mandated for closure.
A thorough understanding of risk ensures that resources are dedicated to the most serious problems. When reporting to management in quality review meetings, good risk management approach also makes it much easier to address the most significant quality events and actions taken. Great advantages of efficiency and effectiveness come as a result of eliminating Death by CAPA.
While procedures such as GHTF CAPA Study Three, ISO 14901, and others provide useful guidelines, an organization must make sure the terms used to describe definitions of impact and frequency in its risk matrix are effective for its particular products and services. Subsequently, these definitions must also make it possible to effectively handle all of the quality events that feed into the system and ensure each event can be dispositioned according to appropriate actions.
Senior management must recognize that daily decisions will be made using these risk scales and must approve of and endorse the dispositioning path. They, not the people following the risk process and executing decisions, will be held accountable.
When first setting up risk definitions and using a new risk matrix, it is also prudent to validate the effectiveness of the system by conducting a post-setup audit. This can easily be achieved by examining quality events after three- and six-months time. These audits allow the organization to thoroughly check the effectiveness of the disposition path and whether or not the appropriate actions (correction, corrective, or preventive) were taken to resolve the quality event. A post-setup audit also allows you to make any necessary adjustments to terms/definitions, review the process with senior management, and monitor and recheck after a few months until it can be refined into the most effective system.
By tracking data generalized over a wide array of device manufacturers, it is evident that the overall number of quality events that have advanced to full investigations and corrective actions have been reduced by more than 50% over the last five years. The industry as a whole has also seen much better utilization of resources against more serious problems. Quality, engineering, manufacturing and development departments, just to name a few, are working much better together, because common goals are realized and focus is aligned to quality assurance and business/production objectives.
Monthly quality review meetings with management have generally become more productive, as key CAPA actions are reviewed and management inputs the decision criteria to the quality team. Conversely, the quality team provides useful data that helps management make better daily decisions that serve long-term goals.
The Current State of CAPA and Beyond
What do we see after 10 years of successes and failures? Many companies are still failing regulatory inspections. As we better understand how to utilize risk and select the appropriate paths and actions, we come to see that the later stages of the CAPA process are often not well executed. The most common failure is poor effectiveness-checking to ensure that a higher degree of risk investigation and corrective response to the root cause has indeed solved the problem and eliminated the risk. The FDA is constantly writing audit findings for this flaw. While the front end of the CAPA process is improving, the back end is not.
The new emphasis in the 2016 international standard will focus more on prevention. This may cause some confusion and an over-emphasis in this area, perhaps bringing on a smaller stage of Death by CAPA for those who do not understand how to deploy or over-deploy this emphasis.
Automation of the CAPA process, when done correctly, can provide a great advantage, and likewise when done poorly can be a significant disadvantage. Many small device manufacturers would be wise to make sure that the CAPA process works on paper and that the procedures align before stepping into a rigorous automated control system. Once aligned, automation can provide many benefits, including document control, reports, management meeting tools, and alerts and alarms for adverse events and other escalations.
The CAPA process, otherwise referred to as deviation management, is a daily system of placing quality events or your company’s problems into a process that takes them from deviation to resolution. Placing continuous quality improvement (CQI) projects into the CAPA process is a recipe for additional problems. If you wish to move your CQI initiatives into a parallel system outside of CAPA, you must have a two-way, or binary, gate where you can only go one way or the other, similar to only being able to turn left or right when encountering a fork in the road. Misunderstanding this binary gate concept is leading to a new wave of Death by CAPA for some organizations.
Conclusion
Companies using a risk system commensurate with the new standards are seeing great advances in resource prioritization, time management, production advances, and many other facets of more productivity and effectiveness. Risk considerations are helping eliminate Death by CAPA and turning the CAPA system into a highly integrated part of the organization for managing and reporting on quality events in a broader quality event management process as part of the overall quality management system.
Moving forward, regulatory agencies will require more of manufacturers in order to demonstrate they are eliminating risky situations from all aspects of the business. Incorporation of these techniques into your systems will bring better results both now and perpetually in the years ahead.
Related Articles
-

Companies can make mistakes during drug and device development and in the post-marketing maintenance phase,…
-

Beginning October 1, 2023, the FDA’s Total Product Life Cycle Advisory Program (TAP) Pilot will…
-

The acquisition expands Veranex’s footprint for product design engineering services across the U.S., with combined…
-

The acquisition brings MAXIS’ expertise in pre-clinical engineering, design validation and verification, regulatory affairs, field…
