UDI: What You Need to Know About Timelines, Compliance and Submissions
While device companies comply with UDI regulations, they also should capitalize on advantages of streamlining procedures in uniquely identifying and improving patient safety, surveillance, and procedures. Medical devices that are presumed critical are subject to this mandate, starting with the most critical (Class III) and ending with least critical (Class I) devices.
Timelines for compliance
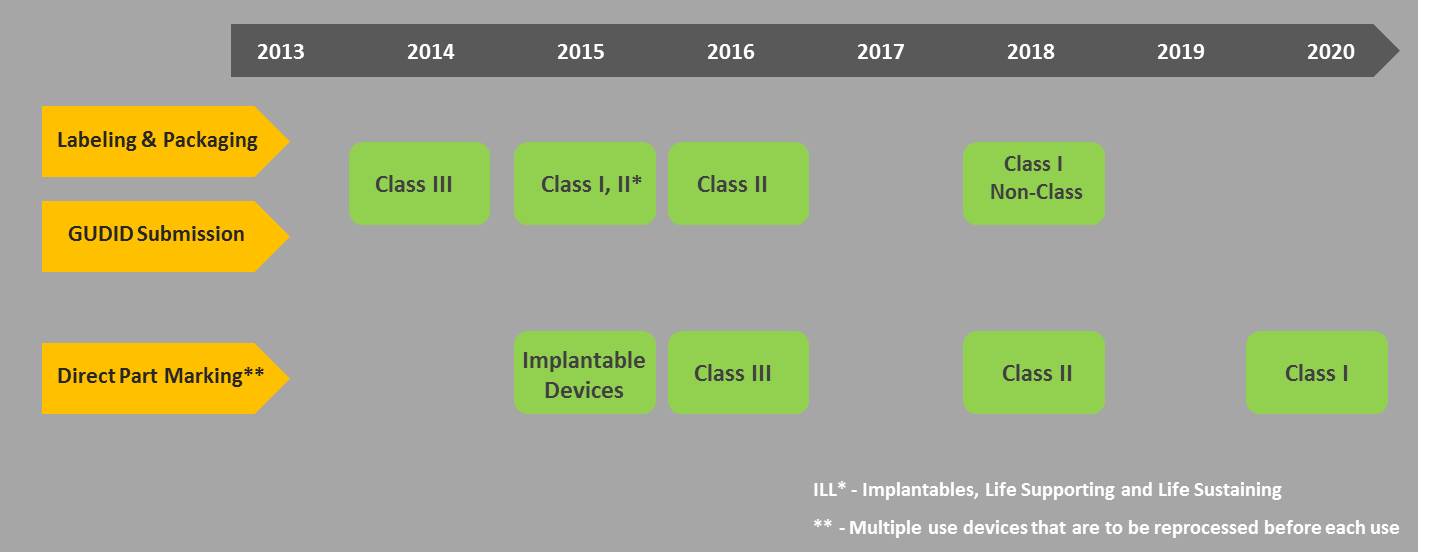
Dates for implementation of the UDI mandate are phased as such where; higher-risk devices are given the first priority i.e. Class III, Class II and Class I.
- Class III devices and devices falling under the PHS act have to be compliant by September 24, 2014. However, an exception was added that a request for a year extension can be made to FDA.
- All implantables, life-supporting, and life-sustaining devices including stand-alone software’s should be bearing a UDI by September 24, 2015.
- All Class III and devices licensed under the PHS Act, for certain intended uses should be directly marked with a UDI.
- All Class II devices should be compliant as well by September 24, 2016.
- All Class I and non-classified devices should be compliant by September 24, 2018.
- Direct Marking of Class II devices for certain intended uses should be compliant by 2018 as well.
- Direct Marking of Class I and unclassified devices, for certain intended uses should be compliant by September 24, 2020.
Implementation challenges
For majority of companies, adoption of UDI in their current existing processes is a major challenge; cost of UDI adoption, re-designing the current technologies, new labeling standards and data mining being the rest of challenges. Most companies feel that the real challenge is a lack of finalized UDI regulation. However, device manufacturers have no choice but to start implementing a comprehensive, compliant, end-to-end solution that is flexible and scalable to handle their increasingly complex product data management needs thereby ultimately bring in UDI compliance. At the end of the day, FDAs sole purpose is to improve patient safety with more accurate reporting and to have a standard procedure where device recalls are made effective and streamlined.
Gearing up
The task for device manufacturer’s is how to comply with the FDA UDI ruling within the assigned timelines. Non-fulfilment will have a major business influence such as devices that are imported would be labeled non-compliant and will not be accepted to enter into the US market. The value of compliance will be greater for manufacturers that export from multiple countries and use different ERPs and PLMs. Implementation of UDI is a critical factor where processes such as Gap analysis, Readiness assessment and Data management should be planned ahead of time to succeed in UDI compliance. UDI is here for a long run and device companies should gear up well ahead of time to avoid any non-compliance issues.
Going global
All device manufacturers have to comply with the UDI mandate. Companies that do not comply with UDI guidelines may face a potential issue of their devices being non-compliant. This may cause impairment to the manufacturer’s brand name and esteem. In addition to USFDA, multiple other countries are planning to follow FDAs footsteps. European Commission already recommended a common framework for a UDI system for medical devices. China has been discussing about implementing a UDI system to track distribution of medical devices for years. Medical device industry experts now suggest that China plans to implement such as system by year end. Health Canada is also actively participating on the UDI framework item that is underway by the International Medical Device Regulators Forum. Members of IMDRF currently include Australia, Brazil, Canada, the European Union, and Japan, as well as the US. Many more emerging global markets may be lost to the manufacturers who are slow in adoption or are unable to comply with these anticipated new global product identification rules.
Issuing agencies
FDA has authorized only three agencies to issue UDIs to the device manufacturers. A FDA accredited Issuing Agency should be operating a system for assignment of UDIs as per the FDA. Currently there are three agencies authorized by the FDA for issuing UDIs: GS1, HIBCC and ICCBBA. Out of the three, GS1 is preferred by most of the companies because of its global approach. GS1 uses the Global Trade Item Number™ (GTIN™) for the unique identification of products worldwide. As per GS1 standards the assigned DI would be of 12 to 14 digits in length, where DIs with less than 14 digits should be appended with leading zeros. Companies with GTINs can also store and access their product data through Global Data Synchronization Network (GDSN), a GS1 data pool network.
The second-most preferred agency is HIBCC, which stands for Health Industry Business Communications Council. HIBCCs codes are alphanumeric and 6 to 23 characters in length. Device companies can create device identifiers by utilizing the HIBC Supplier Labeling Standard (SLS).
The third agency is ICCBBA, that focuses especially on Human, Cell and Tissue-based products like blood bad vendors and other products of human origin.
Achieving UDI compliance

Achieving UDI compliance master data management
The UDI regulation is urging the need for medical device manufacturer’s to redesign their data management strategies to assure that they can provide FDA with accurate device data submissions. All the required medical device data is dispersed across various systems within the organization and may not be available in the necessary format. The required medical device regulatory data should be identified through a well-integrated Master Data Management strategy. The acquired data should be migrated, normalized, collated and validated as per FDA guidelines and specifications. Controlled vocabularies also should be identified and normalized according to FDA specifications.
Format readiness
Preparing the product and regulatory data in the required format is a challenge to the device companies. There are a total of 62 data attributes that are required by FDA, of which 57 are submitted by the device manufacturer and remaining five are auto-populated by the GUDID. FDA will only allow seven days to correct an imperfect submission, which presents the manufacturer with limited choice except for doing the submission right at first time. If perfectly brought into play, the UDI data will be useful in analytics, research studies across various industries, academics, and health authorities, providing a better understanding of device use and achieved patient outcomes.
Technology aspects
All ERPs and PLMs should be updated and modified for UDI compliance as per FDA protocols and UDI issuing agency standards. Software should be installed and well-integrated with the existing systems to manage GUDID compliance. Validation should be done as per current regulatory standards containing project planning, pre-requisites, risk assessment and mitigation, testing protocols and reports. Companies should start implementing solutions to simplify processes, increase productivity and reduce the total cost of compliance. Medical device companies are facing challenges as they add UDI aspects to their products and existing systems. Most of large and medium companies use ERP or PLM applications for lifecycle data management. Costs of implementing such solutions would be a nightmare for small companies.
GUDID
A public database GUDID will serve as the master repository of device identification information. A search option is made available to the public to access device information. GUDID only collects device identifier information; any kind of PI information is not submitted or stored in the GUDID. The presence of a PI attribute is entered in GUDID through a Yes/No option. FDAs GUDID interface is suitable for small companies that have less number of products. Only one device record at a time can be submitted in a manual data entry process. Draft versions of DI record can be saved when using GUDID interface. Device attribute information to the GUDID can be submitted through two ways i.e. either by using FDAs GUDID interface or by HL7 SPL submissions. SPL stands for Structured Product Labeling and is based on standards of HL7. Valid XMLs are created and submitted through FDAs ESG.
The SPL method is suitable for companies having more products. SPL submission method doesn’t have the facility of saving draft records. The device information can be submitted to GUDID either by the device manufacturer or the manufacturer can authorize a third-party person to do it on its behalf after getting approval from the FDA.
Data submission
Regardless of the method chosen for GUDID device information submission, every manufacturer (labeler) should have a GUDID account. A search function is built in GUDID which is free and doesn’t require an account. During search the manufacturer’s details are not disclosed. A labeler organization may have more than one GUDID account. GUDID account is requested by providing a DUNS number after proper verification if the information is correct in Dun and Bradstreet (D&B) database. Complete and send the new GUDID account request form to FDA and once approved the helpdesk will send the login credentials. For GUDID interface users, once the account request is approved, FDA will create a Coordinator account and provide the login credentials to the coordinator. Then the coordinator can create labeler data entry users and assign the labeler DUNS for data entry accordingly. For HL7 SPL submission method, test submissions have to be performed through a GUDID test account. Upon successful test submissions, a request for GUDID production account is made for device data submissions.
Related Articles
-

The FDA does not intend to enforce the GUDID submission requirements for Class I and…
-
As of June 1, 2022, the Center for Devices and Radiological Health plans to accept…
-
Manufacturers preparing premarket submissions should provide performance specifications for the quantitative imaging functions, supporting performance…
-

Taking these compliance challenges into account will allow healthcare organizations to prepare for compliance audits,…
