Biological Evaluation of Medical Devices
The biological safety of various categories of possible toxicological effects should be considered for a particular device. These include; cytotoxicity, irritation, acute systemic toxicity, hemocompatibility and thrombogenicity as short-term effects, and sensitization, genotoxicity, subchronic and chronic toxicity, carcinogenity, and reproduction toxicity as long-term effects. In order to completely evaluate the biological safety of a medical device, the nature and duration of body contact must be considered.
For such a biological safety evaluation, manufacturers most often use the ISO 10993 standard series “Biological evaluation of medical devices”. This standard is internationally accepted; however, many countries have additional requirements or interpret this standard differently.
According to ISO 10993-1 [1], the biological risk of a product should be evaluated and tested within the scope of risk management. The first step of this process is to determine if there is body contact. The standard is not applicable for products with no direct or indirect body contact. For products with direct or indirect body contact, the device components are categorized following ISO 10993-1:2009 for nature of contact (e.g. surface, externally communicating, implant), type of tissue contacted (e.g. skin, mucosal membrane, compromised/breached surfaces, tissue/ bone/dentin, circulating blood), and contact duration (e.g. limited, prolonged, permanent), where the contact duration is the cumulative sum of single, multiple or repeated use contact. Based upon the categorization of the device under evaluation, the manufacturer should develop an appropriate testing strategy. The test strategy should also take into account where the device will be submitted for registration and marketed, e.g. EU, USA, Japan, China, etc., because countries often have specific requirements for the biological evaluation. This two-part article focuses on approval aspects in the European Union (CE Mark) and United States (FDA Approval).
ISO 10993 Standard Series
ISO 10993-1 includes a table (see Table 1 below) that provides “… a framework for the development of an assessment program…” where, for the various intended uses, a general biological test strategy is described. Within the table, the toxicological effects to be considered based on the intended use of the device are marked with an “X”. The manufacturer should consider if data are available which cover the marked biological effects. If the biological effect is determined to be not relevant for this device, or if adequate data are available, further testing in this category is not needed. Thus, an “X” in the aforementioned table does not mean that a test is a required.
However according to ISO 10993-1, additional biological effects such as; chronic toxicity, carcinogenicity, biodegradation, toxicokinetics, immunotoxicity, reproductive/developmental toxicity or other organ-specific toxicities need to be considered depending on the intended use of the device and assessment of risk.
After it is determined which biological effects are relevant and should be considered, the safety risk of each of these effects needs to be addressed. This can be done with testing or a documented justification. A justification can take the place of testing in regulatory submissions when the device components are found to be equivalent to marketed device materials. To document these conclusions, the marketed device materials should be listed and the method of establishing equivalence identified (i.e. manufacturing equivalence memo or comparative analytical chemistry testing).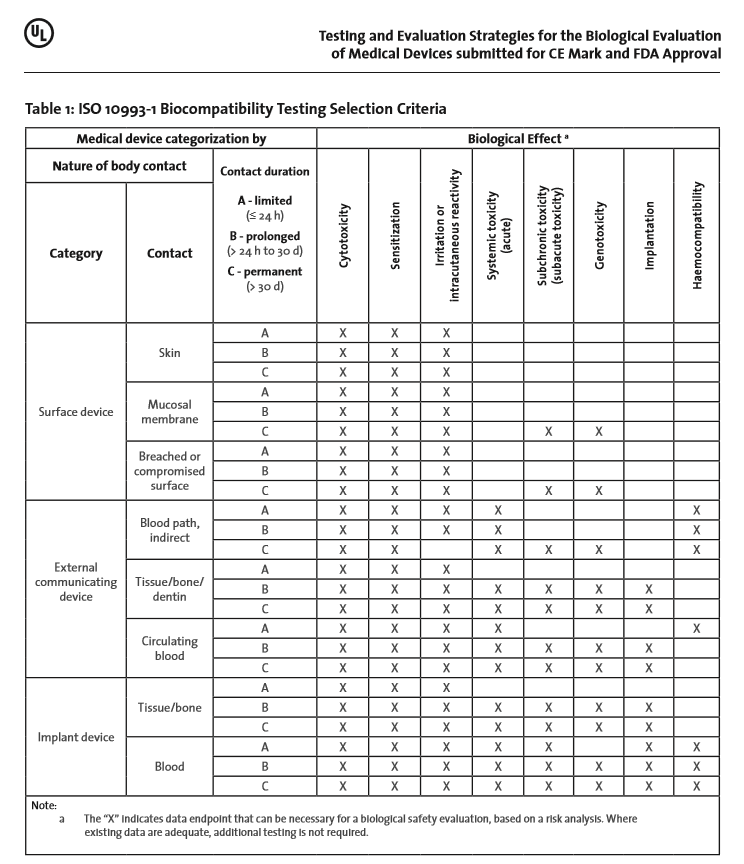
An additional alternative to testing is to use raw materials that are known to be biocompatible and have a long history of safe-use as a medical device material, i.e. stainless steel acc. to ISO 5832-1 [2] / ASTM F139 [3] or Ti6Al4V acc. to ISO 5832-3 [4] / ASTM F 136 [5]. For these materials, it is sufficient to show that removal of process related surface residues at the end of the manufacturing process has been performed.
Ideally, testing should be performed according to the standards recognized by the applicable regulatory bodies. The ISO 10993 series of standards is recognized in both the EU and US. The test methods used for determining various biological effects are described in the individual parts of the ISO 10993 standards. Tests are typically performed using final packaged and sterilized devices, so that all possible effects of the multiple production steps are included in the test sample. However, when the device polymerizes in-situ or is biodegradable, the interim reaction and degradation products must also be investigated for potential biological risk.
In addition to biological tests, the ISO 10993 series includes several standards specifically for physico-chemical tests. The assumption is that the biological effects of a device are dependent not only on the device’s chemical structure, but also on its physical and morphological characteristics. These may have a significant influence on the product’s biocompatibility. For example, a change in the roughness of a surface (change of morphology) may directly influence the cytotoxicity of the material whereas a chemically safe material with rough edges or particulates that can physically harm patient causing irritation.
In order to avoid unnecessary animal testing (requirement of ISO 10993-2), ISO 10993-1 specifies starting the biological evaluation with chemical and physical characterization of the materials (Clause 4.3). Therefore, the final device, including intermediate reaction and degradation products (see above) should be subjected to material characterization as described in ISO 10993-18. It is important to determine the identity and amount of leachable substances that may be bioavailable during the intended use of the device. Data on extractables and leachables should be cross-checked with toxicological databases, such as RTECS and TOXLINE, and an estimation of the benefits vs. risks for the final product determined. Identifying and estimating a potential health hazard early in the design and manufacturing process can avoid unnecessary animal testing, reduce costs and prevent delays in case material changes and retesting is indicated. In the UL white paper, “Physical and Chemical Characterization of Medical Device Materials“ [6] additional background information for applying ISO 10993-18 [7] is described. Furthermore, ISO 10993-1 requires conducting in vitro test procedures before conducting in vivo tests. Also, before starting in vivo tests results of in vitro tests should be available.
Additionally, if the final packed device is sterilized using ethylene oxide, the levels of Ethylene Oxide (EO) and Ethylene Chlorohydrin (ECH) residuals and whether they are in compliance with the limits specified in ISO 10993-7 [8] should be determined.
Next week, in the next part of this article, we will cover Test Strategies for CE Mark, and FDA Approval.
References:
- ISO 10993-1:2009, Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management process
- ISO 5832-1:2007, Implants for surgery – Metallic materials – Part 1: Wrought stainless steel
- ASTM F139:2012, Standard Specification for Wrought 18Chromium-14Nickel-2.5Molybdenum Stainless Steel Sheet and Strip for Surgical Implants (UNS S31673)
- ISO 5832-3:1996, Implants for surgery – Metallic materials – Part 3: Wrought titanium 6-aluminium 4-vanadium alloy
- ASTM F 136:2013, Standard Specification for Wrought Titanium-6Aluminum-4Vanadium ELI (Extra Low Interstitial) Alloy for Surgical Implant Applications (UNS R56401)
- Peeters J (2013) Physical and Chemical Characterization of Medical Device Materials, UL White Paper
- ISO 10993-18:2005, Biological evaluation of medical devices – Part 18: Part 18: Chemical characterization of materials
- ISO 10993-7:2008 Biological evaluation of medical devices – Part 7: Ethylene oxide sterilization residuals
Related Articles
-

The updated guidance clarifies how the program applies to medical devices that may address health…
-

The UK MHRA has published “Software and Artificial Intelligence as a Medical Device.” The guidance…
-

At Device Talks Boston in May, Ronald Kurz, Sr. Director and GM at Canon Virginia,…
-
The FDA is seeking input from organizations interested in collaborating with the agency on the…
